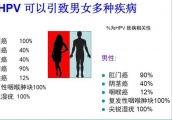
 此前,2017年10月24日,原食品药品监管总局曾公开征求《药品注册管理办法(修订稿)》意见。昨日公布的药品注册管理办法(修订草案征求意见稿)与上次修订稿相比,修改幅度较大。据(修订草案征求意见稿)第一百二十九条,本办法自20**年*月*日起施行。原国家食品药品监督管理局于2007年7月10日公布的《药品注册管理办法》同时废止。下面是赛柏蓝梳理的部分《药品注册管理办法(修订草案征求意见稿)》亮点。药品注册类别细化第五条(药品注册类别)规定,药品注册类别,按照中药、化学药和生物制品等进行分类。中药注册分类:创新药,改良型新药,古代经典名方中药复方制剂,同方类似药,境外已上市境内未上市中药等。化学药注册分类:创新药,改良型新药,仿制药,境外已上市境内未上市化学药等。生物制品注册分类:创新生物制品,改良型生物制品,境内已上市生物制品(包括生物类似药和不按生物类似药管理的境内已上市生物制品),境外已上市境内未上市生物制品等。国家药品监督管理局(以下简称国家局)将根据注册药品的产品特性、创新程度和审评管理需要,组织制定发布各类药品的细化分类和相应的申报资料要求。关联审评审批制度确立第十四条(加快上市注册制度)规定,支持以临床价值为导向的药物创新,建立药品加快上市注册制度。对符合条件的药品注册申请,申请人可申请进入突破性治疗药物程序、附条件批准程序、优先审评审批程序及特别审批程序,在药品研制和注册过程中,给予技术指导、全过程沟通、优先配置资源、缩短审评时限等政策支持。第十五条(关联审评审批制度)规定,建立关联审评审批制度。在审批药品时,对化学原料药一并审评审批,对相关辅料、直接接触药品的包装材料和容器一并审评。其中,化学原料药在药品制剂审批时一并审批;国内已上市药品的化学原料药可申请单独审评。建立化学原料药、相关辅料及直接接触药品的包装材料和容器(以下简称原辅包)信息登记平台,对原辅包信息进行登记,在药品制剂审评时关联。大力支持中药传承第二十条(支持中药传承创新)规定,支持中药传承和创新。建立完善符合中药特点的注册管理制度和技术评价体系,将中药传统优势与药品研发科学要求相结合。中药创新药,应当突出疗效新的特点;中药改良型新药,应当体现临床应用优势;经典名方类中药,按照简化标准审评审批;天然药物,按照现代医学标准审评审批。提高中药临床研究能力,中药注册申请需提交临床价值和资源评估材料,突出以临床价值为导向,促进资源可持续利用。鼓励运用现代科学技术研究开发传统中成药,鼓励发挥中药传统剂型优势研制中药新药,加强中药质量控制。在起草说明中,国家药监局表示为做好中药传承创新,建立和完善符合中药特点技术评价体系,国家药监局正在研究制定专门的管理制度,将以配套文件形式单独发布。同步进行GMP检查药品批准上市前需要同步进行GMP检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查。省级局在完成GMP检查后应当将GMP检查情况、检查结果等相关材料反馈国家局药品审评中心进行综合审评。第四十九条(生产现场检查与GMP衔接)规定,对于创新药、改良型新药以及按新药管理的生物制品等,应当进行药品注册生产现场检查。申请人在接到药品注册生产现场检查时,应当向所在地省级局提出同步进行GMP检查;已获得相应生产范围生产许可证的,可不提出进行GMP检查。对于未获得相应生产范围药品生产许可证的仿制药等,由国家局药品审评中心基于风险启动药品注册生产现场检查。启动药品注册生产现场检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查;不需要启动药品注册生产现场检查的,国家局药品审评中心通知申请人向所在地省级局提出进行GMP检查。对于已获得相应生产范围药品生产许可证且已有同剂型品种上市的仿制药等,一般不再进行药品注册生产现场检查。药品生产工艺变更补充申请第五十九条(核准程序)规定,药品生产工艺、质量标准、说明书和标签,由申请人在提出药品注册申请时提出,国家局药品审评中心在审评时予以核准,批准药品注册申请时发给申请人。经核准的药品生产工艺、质量标准、说明书和标签纳入药品品种档案,并根据上市后变更情况及时更新。药品生产工艺、质量标准、说明书和标签的通用格式和撰写指南,由国家局药品审评中心另行制定,并向社会公布。第六十条(生产工艺) 药品批准上市后,持有人应按照国家局核准的生产工艺生产药品,并根据《药品生产质量管理规范》要求进行细化和实施。变更生产工艺的,持有人应当在实施前进行充分研究和验证,并按照本办法有关药品上市后变更的程序提出补充申请、备案或者报告。多类药品优先审评审批第七十二条(优先范围)规定,药品上市注册申请时,以下具有明显临床价值的药品,可申请进入优先审评审批程序:(一)纳入突破性治疗药物程序的药品;(二)纳入附条件批准上市注册的药品;(三)临床急需的短缺药品、防治重大传染病和罕见病等疾病的新药;(四)符合儿童生理特征的儿童用药品新品种、剂型和规格;(五)疾病预防、控制急需的疫苗和创新疫苗;(六)其他可以实行优先审评审批的情形。第七十四条(支持政策)规定,对纳入优先审评审批程序的药品上市注册申请,给予以下政策支持:(一)药品上市注册审评时限120个工作日;(二)临床急需的境外已上市的罕见病药品审评时限60个工作日;(三)需要检查检验的,优先安排检查检验;(四)按要求滚动提交资料。欲查看《药品注册管理办法(修订草案征求意见稿)》原文,请点击文末左下角阅读原文。
此前,2017年10月24日,原食品药品监管总局曾公开征求《药品注册管理办法(修订稿)》意见。昨日公布的药品注册管理办法(修订草案征求意见稿)与上次修订稿相比,修改幅度较大。据(修订草案征求意见稿)第一百二十九条,本办法自20**年*月*日起施行。原国家食品药品监督管理局于2007年7月10日公布的《药品注册管理办法》同时废止。下面是赛柏蓝梳理的部分《药品注册管理办法(修订草案征求意见稿)》亮点。药品注册类别细化第五条(药品注册类别)规定,药品注册类别,按照中药、化学药和生物制品等进行分类。中药注册分类:创新药,改良型新药,古代经典名方中药复方制剂,同方类似药,境外已上市境内未上市中药等。化学药注册分类:创新药,改良型新药,仿制药,境外已上市境内未上市化学药等。生物制品注册分类:创新生物制品,改良型生物制品,境内已上市生物制品(包括生物类似药和不按生物类似药管理的境内已上市生物制品),境外已上市境内未上市生物制品等。国家药品监督管理局(以下简称国家局)将根据注册药品的产品特性、创新程度和审评管理需要,组织制定发布各类药品的细化分类和相应的申报资料要求。关联审评审批制度确立第十四条(加快上市注册制度)规定,支持以临床价值为导向的药物创新,建立药品加快上市注册制度。对符合条件的药品注册申请,申请人可申请进入突破性治疗药物程序、附条件批准程序、优先审评审批程序及特别审批程序,在药品研制和注册过程中,给予技术指导、全过程沟通、优先配置资源、缩短审评时限等政策支持。第十五条(关联审评审批制度)规定,建立关联审评审批制度。在审批药品时,对化学原料药一并审评审批,对相关辅料、直接接触药品的包装材料和容器一并审评。其中,化学原料药在药品制剂审批时一并审批;国内已上市药品的化学原料药可申请单独审评。建立化学原料药、相关辅料及直接接触药品的包装材料和容器(以下简称原辅包)信息登记平台,对原辅包信息进行登记,在药品制剂审评时关联。大力支持中药传承第二十条(支持中药传承创新)规定,支持中药传承和创新。建立完善符合中药特点的注册管理制度和技术评价体系,将中药传统优势与药品研发科学要求相结合。中药创新药,应当突出疗效新的特点;中药改良型新药,应当体现临床应用优势;经典名方类中药,按照简化标准审评审批;天然药物,按照现代医学标准审评审批。提高中药临床研究能力,中药注册申请需提交临床价值和资源评估材料,突出以临床价值为导向,促进资源可持续利用。鼓励运用现代科学技术研究开发传统中成药,鼓励发挥中药传统剂型优势研制中药新药,加强中药质量控制。在起草说明中,国家药监局表示为做好中药传承创新,建立和完善符合中药特点技术评价体系,国家药监局正在研究制定专门的管理制度,将以配套文件形式单独发布。同步进行GMP检查药品批准上市前需要同步进行GMP检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查。省级局在完成GMP检查后应当将GMP检查情况、检查结果等相关材料反馈国家局药品审评中心进行综合审评。第四十九条(生产现场检查与GMP衔接)规定,对于创新药、改良型新药以及按新药管理的生物制品等,应当进行药品注册生产现场检查。申请人在接到药品注册生产现场检查时,应当向所在地省级局提出同步进行GMP检查;已获得相应生产范围生产许可证的,可不提出进行GMP检查。对于未获得相应生产范围药品生产许可证的仿制药等,由国家局药品审评中心基于风险启动药品注册生产现场检查。启动药品注册生产现场检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查;不需要启动药品注册生产现场检查的,国家局药品审评中心通知申请人向所在地省级局提出进行GMP检查。对于已获得相应生产范围药品生产许可证且已有同剂型品种上市的仿制药等,一般不再进行药品注册生产现场检查。药品生产工艺变更补充申请第五十九条(核准程序)规定,药品生产工艺、质量标准、说明书和标签,由申请人在提出药品注册申请时提出,国家局药品审评中心在审评时予以核准,批准药品注册申请时发给申请人。经核准的药品生产工艺、质量标准、说明书和标签纳入药品品种档案,并根据上市后变更情况及时更新。药品生产工艺、质量标准、说明书和标签的通用格式和撰写指南,由国家局药品审评中心另行制定,并向社会公布。第六十条(生产工艺) 药品批准上市后,持有人应按照国家局核准的生产工艺生产药品,并根据《药品生产质量管理规范》要求进行细化和实施。变更生产工艺的,持有人应当在实施前进行充分研究和验证,并按照本办法有关药品上市后变更的程序提出补充申请、备案或者报告。多类药品优先审评审批第七十二条(优先范围)规定,药品上市注册申请时,以下具有明显临床价值的药品,可申请进入优先审评审批程序:(一)纳入突破性治疗药物程序的药品;(二)纳入附条件批准上市注册的药品;(三)临床急需的短缺药品、防治重大传染病和罕见病等疾病的新药;(四)符合儿童生理特征的儿童用药品新品种、剂型和规格;(五)疾病预防、控制急需的疫苗和创新疫苗;(六)其他可以实行优先审评审批的情形。第七十四条(支持政策)规定,对纳入优先审评审批程序的药品上市注册申请,给予以下政策支持:(一)药品上市注册审评时限120个工作日;(二)临床急需的境外已上市的罕见病药品审评时限60个工作日;(三)需要检查检验的,优先安排检查检验;(四)按要求滚动提交资料。欲查看《药品注册管理办法(修订草案征求意见稿)》原文,请点击文末左下角阅读原文。 最新| 国家发文,大力支持中药传承
作者:赛柏蓝新媒体 2019-10-04阅读:2390次
▍▍整理:赛柏蓝-遥望国家药监局发布《药品注册管理办法》(修订草案征求意见稿),提出大力支持中药传承。昨日(9月30日),国家药监局再发重磅文件征求意见稿——涉及药品注册、生产监督、经营监督,征求意见稿的意见征集时间为期一个月。在这些管理办法后,一系列配套支持文件也将陆续出台。有医药界权威人士向赛柏蓝表示,此次,国家药监局发布的三份征求意见稿,是医药行业最重要的政策。此前,2017年10月24日,原食品药品监管总局曾公开征求《药品注册管理办法(修订稿)》意见。昨日公布的药品注册管理办法(修订草案征求意见稿)与上次修订稿相比,修改幅度较大。据(修订草案征求意见稿)第一百二十九条,本办法自20**年*月*日起施行。原国家食品药品监督管理局于2007年7月10日公布的《药品注册管理办法》同时废止。下面是赛柏蓝梳理的部分《药品注册管理办法(修订草案征求意见稿)》亮点。药品注册类别细化第五条(药品注册类别)规定,药品注册类别,按照中药、化学药和生物制品等进行分类。中药注册分类:创新药,改良型新药,古代经典名方中药复方制剂,同方类似药,境外已上市境内未上市中药等。化学药注册分类:创新药,改良型新药,仿制药,境外已上市境内未上市化学药等。生物制品注册分类:创新生物制品,改良型生物制品,境内已上市生物制品(包括生物类似药和不按生物类似药管理的境内已上市生物制品),境外已上市境内未上市生物制品等。国家药品监督管理局(以下简称国家局)将根据注册药品的产品特性、创新程度和审评管理需要,组织制定发布各类药品的细化分类和相应的申报资料要求。关联审评审批制度确立第十四条(加快上市注册制度)规定,支持以临床价值为导向的药物创新,建立药品加快上市注册制度。对符合条件的药品注册申请,申请人可申请进入突破性治疗药物程序、附条件批准程序、优先审评审批程序及特别审批程序,在药品研制和注册过程中,给予技术指导、全过程沟通、优先配置资源、缩短审评时限等政策支持。第十五条(关联审评审批制度)规定,建立关联审评审批制度。在审批药品时,对化学原料药一并审评审批,对相关辅料、直接接触药品的包装材料和容器一并审评。其中,化学原料药在药品制剂审批时一并审批;国内已上市药品的化学原料药可申请单独审评。建立化学原料药、相关辅料及直接接触药品的包装材料和容器(以下简称原辅包)信息登记平台,对原辅包信息进行登记,在药品制剂审评时关联。大力支持中药传承第二十条(支持中药传承创新)规定,支持中药传承和创新。建立完善符合中药特点的注册管理制度和技术评价体系,将中药传统优势与药品研发科学要求相结合。中药创新药,应当突出疗效新的特点;中药改良型新药,应当体现临床应用优势;经典名方类中药,按照简化标准审评审批;天然药物,按照现代医学标准审评审批。提高中药临床研究能力,中药注册申请需提交临床价值和资源评估材料,突出以临床价值为导向,促进资源可持续利用。鼓励运用现代科学技术研究开发传统中成药,鼓励发挥中药传统剂型优势研制中药新药,加强中药质量控制。在起草说明中,国家药监局表示为做好中药传承创新,建立和完善符合中药特点技术评价体系,国家药监局正在研究制定专门的管理制度,将以配套文件形式单独发布。同步进行GMP检查药品批准上市前需要同步进行GMP检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查。省级局在完成GMP检查后应当将GMP检查情况、检查结果等相关材料反馈国家局药品审评中心进行综合审评。第四十九条(生产现场检查与GMP衔接)规定,对于创新药、改良型新药以及按新药管理的生物制品等,应当进行药品注册生产现场检查。申请人在接到药品注册生产现场检查时,应当向所在地省级局提出同步进行GMP检查;已获得相应生产范围生产许可证的,可不提出进行GMP检查。对于未获得相应生产范围药品生产许可证的仿制药等,由国家局药品审评中心基于风险启动药品注册生产现场检查。启动药品注册生产现场检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查;不需要启动药品注册生产现场检查的,国家局药品审评中心通知申请人向所在地省级局提出进行GMP检查。对于已获得相应生产范围药品生产许可证且已有同剂型品种上市的仿制药等,一般不再进行药品注册生产现场检查。药品生产工艺变更补充申请第五十九条(核准程序)规定,药品生产工艺、质量标准、说明书和标签,由申请人在提出药品注册申请时提出,国家局药品审评中心在审评时予以核准,批准药品注册申请时发给申请人。经核准的药品生产工艺、质量标准、说明书和标签纳入药品品种档案,并根据上市后变更情况及时更新。药品生产工艺、质量标准、说明书和标签的通用格式和撰写指南,由国家局药品审评中心另行制定,并向社会公布。第六十条(生产工艺) 药品批准上市后,持有人应按照国家局核准的生产工艺生产药品,并根据《药品生产质量管理规范》要求进行细化和实施。变更生产工艺的,持有人应当在实施前进行充分研究和验证,并按照本办法有关药品上市后变更的程序提出补充申请、备案或者报告。多类药品优先审评审批第七十二条(优先范围)规定,药品上市注册申请时,以下具有明显临床价值的药品,可申请进入优先审评审批程序:(一)纳入突破性治疗药物程序的药品;(二)纳入附条件批准上市注册的药品;(三)临床急需的短缺药品、防治重大传染病和罕见病等疾病的新药;(四)符合儿童生理特征的儿童用药品新品种、剂型和规格;(五)疾病预防、控制急需的疫苗和创新疫苗;(六)其他可以实行优先审评审批的情形。第七十四条(支持政策)规定,对纳入优先审评审批程序的药品上市注册申请,给予以下政策支持:(一)药品上市注册审评时限120个工作日;(二)临床急需的境外已上市的罕见病药品审评时限60个工作日;(三)需要检查检验的,优先安排检查检验;(四)按要求滚动提交资料。欲查看《药品注册管理办法(修订草案征求意见稿)》原文,请点击文末左下角阅读原文。
此前,2017年10月24日,原食品药品监管总局曾公开征求《药品注册管理办法(修订稿)》意见。昨日公布的药品注册管理办法(修订草案征求意见稿)与上次修订稿相比,修改幅度较大。据(修订草案征求意见稿)第一百二十九条,本办法自20**年*月*日起施行。原国家食品药品监督管理局于2007年7月10日公布的《药品注册管理办法》同时废止。下面是赛柏蓝梳理的部分《药品注册管理办法(修订草案征求意见稿)》亮点。药品注册类别细化第五条(药品注册类别)规定,药品注册类别,按照中药、化学药和生物制品等进行分类。中药注册分类:创新药,改良型新药,古代经典名方中药复方制剂,同方类似药,境外已上市境内未上市中药等。化学药注册分类:创新药,改良型新药,仿制药,境外已上市境内未上市化学药等。生物制品注册分类:创新生物制品,改良型生物制品,境内已上市生物制品(包括生物类似药和不按生物类似药管理的境内已上市生物制品),境外已上市境内未上市生物制品等。国家药品监督管理局(以下简称国家局)将根据注册药品的产品特性、创新程度和审评管理需要,组织制定发布各类药品的细化分类和相应的申报资料要求。关联审评审批制度确立第十四条(加快上市注册制度)规定,支持以临床价值为导向的药物创新,建立药品加快上市注册制度。对符合条件的药品注册申请,申请人可申请进入突破性治疗药物程序、附条件批准程序、优先审评审批程序及特别审批程序,在药品研制和注册过程中,给予技术指导、全过程沟通、优先配置资源、缩短审评时限等政策支持。第十五条(关联审评审批制度)规定,建立关联审评审批制度。在审批药品时,对化学原料药一并审评审批,对相关辅料、直接接触药品的包装材料和容器一并审评。其中,化学原料药在药品制剂审批时一并审批;国内已上市药品的化学原料药可申请单独审评。建立化学原料药、相关辅料及直接接触药品的包装材料和容器(以下简称原辅包)信息登记平台,对原辅包信息进行登记,在药品制剂审评时关联。大力支持中药传承第二十条(支持中药传承创新)规定,支持中药传承和创新。建立完善符合中药特点的注册管理制度和技术评价体系,将中药传统优势与药品研发科学要求相结合。中药创新药,应当突出疗效新的特点;中药改良型新药,应当体现临床应用优势;经典名方类中药,按照简化标准审评审批;天然药物,按照现代医学标准审评审批。提高中药临床研究能力,中药注册申请需提交临床价值和资源评估材料,突出以临床价值为导向,促进资源可持续利用。鼓励运用现代科学技术研究开发传统中成药,鼓励发挥中药传统剂型优势研制中药新药,加强中药质量控制。在起草说明中,国家药监局表示为做好中药传承创新,建立和完善符合中药特点技术评价体系,国家药监局正在研究制定专门的管理制度,将以配套文件形式单独发布。同步进行GMP检查药品批准上市前需要同步进行GMP检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查。省级局在完成GMP检查后应当将GMP检查情况、检查结果等相关材料反馈国家局药品审评中心进行综合审评。第四十九条(生产现场检查与GMP衔接)规定,对于创新药、改良型新药以及按新药管理的生物制品等,应当进行药品注册生产现场检查。申请人在接到药品注册生产现场检查时,应当向所在地省级局提出同步进行GMP检查;已获得相应生产范围生产许可证的,可不提出进行GMP检查。对于未获得相应生产范围药品生产许可证的仿制药等,由国家局药品审评中心基于风险启动药品注册生产现场检查。启动药品注册生产现场检查的,申请人在接到药品注册生产现场检查通知时,应当向所在地省级局提出同步进行GMP检查;不需要启动药品注册生产现场检查的,国家局药品审评中心通知申请人向所在地省级局提出进行GMP检查。对于已获得相应生产范围药品生产许可证且已有同剂型品种上市的仿制药等,一般不再进行药品注册生产现场检查。药品生产工艺变更补充申请第五十九条(核准程序)规定,药品生产工艺、质量标准、说明书和标签,由申请人在提出药品注册申请时提出,国家局药品审评中心在审评时予以核准,批准药品注册申请时发给申请人。经核准的药品生产工艺、质量标准、说明书和标签纳入药品品种档案,并根据上市后变更情况及时更新。药品生产工艺、质量标准、说明书和标签的通用格式和撰写指南,由国家局药品审评中心另行制定,并向社会公布。第六十条(生产工艺) 药品批准上市后,持有人应按照国家局核准的生产工艺生产药品,并根据《药品生产质量管理规范》要求进行细化和实施。变更生产工艺的,持有人应当在实施前进行充分研究和验证,并按照本办法有关药品上市后变更的程序提出补充申请、备案或者报告。多类药品优先审评审批第七十二条(优先范围)规定,药品上市注册申请时,以下具有明显临床价值的药品,可申请进入优先审评审批程序:(一)纳入突破性治疗药物程序的药品;(二)纳入附条件批准上市注册的药品;(三)临床急需的短缺药品、防治重大传染病和罕见病等疾病的新药;(四)符合儿童生理特征的儿童用药品新品种、剂型和规格;(五)疾病预防、控制急需的疫苗和创新疫苗;(六)其他可以实行优先审评审批的情形。第七十四条(支持政策)规定,对纳入优先审评审批程序的药品上市注册申请,给予以下政策支持:(一)药品上市注册审评时限120个工作日;(二)临床急需的境外已上市的罕见病药品审评时限60个工作日;(三)需要检查检验的,优先安排检查检验;(四)按要求滚动提交资料。欲查看《药品注册管理办法(修订草案征求意见稿)》原文,请点击文末左下角阅读原文。 
越睡不着越焦虑,越焦虑越睡不着,如何远离失眠的痛苦?
祝你早日康复 · 2018-03-15
身体是这样一步步衰老的,8个症状早就提醒过你
生命时报 · 2018-02-28
注重对外阴的科学洗护才能更健康,哪些洗护不科学你知道吗?
茵茵生活顾问 · 2018-01-10
这5种瑜伽手印改善健康,你知道几个?
瑜伽路上 · 2017-09-21
28岁孕妇怀孕四个月肚子大的像八个月,孩子出生后老公瘫倒在地
Baby爱妈咪 · 2017-09-06
为什么有的孕妈没能保住孩子?子宫5大常见杀手,排第一的直接导致流产!
书籍中的灵魂 · 2017-07-11
蔡依林穿泳衣上街 白癜风患者应该怎么办
买菜的老王 · 2017-06-29