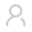

神外前沿讯,3月9日,《癌基因》期刊在线发表了中科院神经所、脑科学与智能技术卓越创新中心、神经科学国家重点实验室熊志奇研究组题为《抑制KPNB1扰乱蛋白质稳态并诱发非折叠蛋白反应介导的胶质母细胞瘤细胞凋亡》的研究文章。
详细揭示了抑制蛋白核转运受体KPNB1产生蛋白稳态失调和非折叠蛋白反应(unfolded protein response, UPR),阐明了凋亡的上游分子信号通路和耐受机制,而相应的联合用药具有更好地肿瘤杀伤效果。
恶性胶质瘤是原发性恶性脑肿瘤最常见的分型。目前的常规治疗手段无显著改善作用。深入了解胶质母细胞瘤(GBM)的致病分子机制有助于发现药物靶标,开发药物和用药策略。KPNB1的表达与胶质瘤的恶性级别以及患者生存率正相关,是潜在的致病基因。KPNB1介导蛋白质的入核,也参与有丝分裂,这对肿瘤细胞的恶性增殖是必须的,故KPNB1是潜在的治疗靶标。科学家已发现抑制KPNB1可诱导肿瘤细胞凋亡,但相关分子机制尚不清楚。

(图注:左上图为科学问题。左下图为研究成果的概要图。右图为抑制KPNB1诱导胶质瘤凋亡的分子机制。)
本项工作中,研究人员发现,抑制KPNB1会下调GBM细胞的生长并诱导凋亡,但不引起有丝分裂障碍。抑制KPNB1上调促凋亡Bcl-2家族蛋白Puma和Noxa,解除抗凋亡Bcl-2家族蛋白Mcl-1和Bcl-xL对凋亡的抑制。据此联用KPNB1抑制剂和Bcl-xL抑制剂能加剧凋亡。
研究者推测,KPNB1的货物蛋白众多,抑制KPNB1或许引起蛋白质错误分布和蛋白稳态失调,产生应激反应。进一步的研究显示,抑制KPNB1使货物蛋白NF-κB p65阻滞在细胞浆中并泛素化,促进多种分子伴侣和自噬相关蛋白与p65结合,并引起泛素化蛋白聚集,激活UPR中的PERK/eIF2α/ATF4通路,上调Puma和Noxa。这支持了上述推测。
此时,细胞通过UPR减少蛋白质合成,并通过自噬和蛋白酶体途径降解蛋白质,以此缓解蛋白过载和细胞毒性,如蛋白质稳态无法恢复,过度激活的UPR即上调促凋亡Bcl-2蛋白,诱导凋亡。溶酶体或蛋白酶体抑制剂加剧蛋白过载,促进KPNB1抑制剂诱导GBM细胞凋亡。
本项工作揭示了KPNB1在维持肿瘤细胞蛋白质稳态中的重要作用,系统地展示了靶向KPNB1引起GBM细胞凋亡和耐受凋亡诱导的分子机制,为后续人们开发KPNB1抑制剂和其他抗肿瘤药联合治疗策略提供了思路和依据,为深入研究GBM的病理和治疗干预方法奠定了坚实的基础。
该项研究主要由博士后朱志川在熊志奇研究员的指导下完成。课题组的其他成员积极参与。本工作得到中国科学院战略性先导科技专项、国家自然科学基金和科技部项目等资助。
图文来源:中科院











