导读
免疫治疗干预已经在各种肿瘤中显示出疗效,但仅有一部分患者出现临床治疗反应。肿瘤微环境中,局部CD8+T细胞免疫应答有利于检查点阻断治疗。然而,随着对有/无产生CD8+T细胞浸润肿瘤分子研究的深入,越来越多的证据表明,肿瘤细胞中的致癌通路激活可以削弱诱导或执行局部抗肿瘤免疫应答。
免疫治疗已经成为各种肿瘤的重要治疗方式。其中,靶向T细胞抑制受体的抗体CTLA-4和PD-1/PD-L1抗体是临床上最先进的抗体,获FDA批准应用于黑色素瘤,NSCLC,肾细胞癌,膀胱癌,霍奇金淋巴瘤,头颈癌,默克尔细胞癌,MSI高肿瘤,肝细胞癌和胃食管结合部癌的治疗。然而,据目前分析,仅有少数肿瘤患者获益。大量研究表明,致癌通路影响局部抗肿瘤免疫反应,致癌通路会通过癌基因获得性功能改变或肿瘤抑制基因功能丧失被激活。
获得性功能改变
致癌性WNT-β-连环蛋白信号传导导致T细胞募集减少。转移性黑色素瘤研究表明,34.2%(91/266例)的黑色素瘤转移病灶是非T细胞发炎的。而在非T细胞炎性病灶中,48%的病灶肿瘤细胞中WNT-β-连环蛋白信号通路被激活。同样地,BRAF突变型原发性黑色素瘤病灶分析显示,浸润性T细胞减少与肿瘤细胞中β-连环蛋白信号上调以及治疗抵抗相关。
为确定肿瘤细胞内在β-连环蛋白致癌性激活与T细胞排斥是否存在因果关系,一项研究将表达Braf V600E和floxed Pten等位基因的基因工程小鼠模型(GEMM)与表达Cre诱导的β-连环蛋白的主要稳定形式的GEMM进行杂交。结果表明,β-连环蛋白阳性和β-连环蛋白阴性BrafLSL−V600E/Ptenfl/fl GEMM均发展为具有100%外显率的黑色素瘤。然而,进一步分析表明,β-连环蛋白阳性肿瘤T细胞浸润最少,并且对检查点阻断剂治疗具有抵抗性。
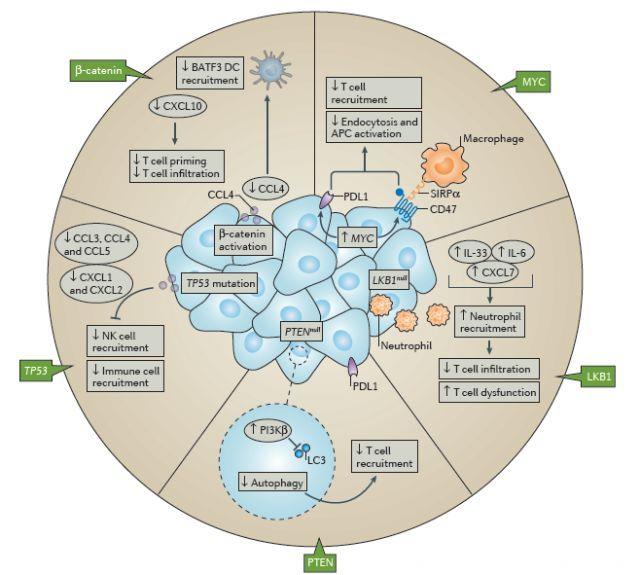
事实上,β-连环蛋白阳性肿瘤CCL4和其它黑色素瘤细胞趋化因子产量减少,这至少是引起BATF3 DC募集至TME中数量严重减少的部分原因,这将导致宿主抗原性T细胞启动缺陷,引起检查点阻断剂治疗耐药性。然而,将成熟的DC注射入β-连环蛋白阳性肿瘤中可以逆转耐药性。总之,这些数据表明,β-连环蛋白信号传导的肿瘤细胞内在激活可能有助于非T细胞炎性肿瘤免疫微环境(TME),产生检查点阻断剂治疗耐药性。
进一步研究表明,β-连环蛋白阳性肿瘤效应T细胞浸润减少也是效应T细胞转运受损所致。因此,β-连环蛋白阳性肿瘤中BATF3 DC缺失导致早期T细胞启动缺陷,以及效应T细胞向TME转移缺陷。这些数据表明,β-连环蛋白上调和BATF3 DC募集缺陷可能介导免疫治疗的继发性耐药。
膀胱癌和头颈癌研究表明,WNT-β-连环蛋白通路可能与黑色素瘤以外其它肿瘤的非T细胞炎性TME相关。此外,正在进行的关于所有实体瘤的TCGA数据分析表明,大多数肿瘤细胞中非T细胞炎性表型和β-连环蛋白信号通路获得性功能也存在类似关联。
MYC获得功能抑制T细胞活化和浸润。转录因子MYC调节细胞增殖,分化和存活,并在多种肿瘤中过表达。通路被激活通常通过基因扩增或组成型表达,而非点突变而发生。一项应用Myc过表达导致骨肉瘤发展的GEMM的研究表明,Myc持续过度表达是肿瘤细胞持久性的必要因素,这种现象称为“致癌基因成瘾”。对同一模型的研究发现,Myc失活时,肿瘤细胞发生细胞周期停滞和细胞凋亡,增加动物存活率。值得注意的是,随后的研究表明,Myc失活后,宿主T细胞可以显著促进肿瘤细胞消除。特别地,虽然Myc重新激活后,诱导细胞周期停滞不需要CD4+T细胞参与,但是诱导衰老必须有CD4+T细胞参与。
从机制上讲,Myc失活导致PD-L1和 白细胞表面抗原CD47表达显著降低。PD-L1和CD47转录直接受MYC控制,通过转染恢复CD47或PD-L1表达可以逆转表型,预防肿瘤相关T细胞积累。CD47或PD-L1表达恢复也会引起Myc失活肿瘤中CD31 +微血管的稳定性以及TIE2和ANG1表达。
功能丧失改变
LKB1功能丧失减少T细胞浸润。除T细胞启动缺陷外,免疫逃逸机制可能还与某些肿瘤内在通路有关。例如,约30%的非小细胞肺癌患者发生肿瘤抑制基因LKB1突变,而LKB1与较差的预后有关。Kras驱动的NSCLC GEMM研究表明,LKB1缺陷型肿瘤中的肿瘤相关嗜中性粒细胞显著增加,同时浸润性T细胞数量适当减少。
PTEN缺失会削弱有效T细胞启动。PTEN失活性突变或缺失与黑色素瘤种T细胞浸润缺陷有关。研究表明,经PD-1抗体治疗的PTEN高表达黑色素瘤患者的临床反应更好。PTEN突变或缺失是一种免疫逃逸机制。目前PTEN缺失或PI3K激活促进免疫逃逸的机制尚不完全清楚。
p53功能丧失减少T细胞浸润。肿瘤抑制基因Trp53失活性突变与免疫渗透减少有关。一项研究表明,肿瘤消退与肿瘤细胞中免疫基因表达增加有关。这些基因包括CSF1,CCL2,CXCL1和IL-15以及ICAM1和VCAM1。在应用相同模型的独立研究中,Trp53诱导引起CCL2,CCL3,CCL4,CCL5,CXCL1和CXCL2以及细胞因子IL-1β,IL-12β和IL-15表达增加。最新人类肿瘤研究也表明,p53状态与免疫细胞浸润具有相关性。
其它致癌通路的影响
早期数据表明,其它致癌通路也可能影响抗肿瘤免疫反应。最新胶质瘤患者以及临床前胶质瘤小鼠模型研究表证实,编码异柠檬酸脱氢酶1和2(IDH1和IDH2)的基因突变与T细胞浸润减少有关。IDH1活性增加导致STAT1蛋白水平降低以及磷酸化STAT1减少。神经胶质瘤细胞中STAT1信号减弱与CXCL10表达减少有关,而CXCL10是效应T细胞募集的关键趋化因子。然而,JAK1-STAT1信号传导与抗PD-1抗体耐药之间是否存在直接联系,以及JAK1-STAT1信号传导是否与肿瘤致癌通路相互干扰仍不清楚。
与STAT1信号传导相反,人类肿瘤中的STAT3信号传导经常被激活。体外研究表明,抑制STAT3信号传导可以产生促炎环境,而增强STAT3信号传导可以降低促炎介质,如趋化因子CCL5和CXCL10表达。基因诱导的前列腺癌模型和致癌物诱导的NSCLC模型研究表明,致癌STAT3信号传导可以调节TME的免疫生物学特性。Pten缺失驱动的前列腺癌模型中,肿瘤细胞发生衰老,并且磷酸化的STAT3增加。STAT3的转录活性诱导趋化因子和细胞因子表达,如CXCL1, CXCL2和巨噬细胞集落刺激因子(MCSF),它们与骨髓来源的抑制细胞(MDSC)募集有关。STAT3信号传导消除或抑制取消了MDSC积累,并恢复肿瘤内T细胞积聚。
此外,最新研究表明,膀胱癌中PPARγ信号传导活化与编码PPARγ的辅因子核激素受体基因维甲酸X受体α(RXRA)突变有关。核因子-κB(NF-κB)信号通路对肿瘤发生和与宿主基质细胞的相互作用具有多种影响。
文章编译自:Impact of oncogenic pathways on evasion of antitumour immune responses.Stefani Spranger1 and Thomas F. Gajewski.1. Nature Reviews Cancer.Jan 2018.
如何使KIR原形毕露?|肿瘤免疫学院IO秒懂系列18









